 |
|
News and Publications
Computational Structural Biology
W. Wriggers, P. Chacón, Y. Cong, J. Kovacs, E. Metwally, S. Birmanns*
* John von Neumann Institut für Computing, Jülich, Germany
COMBINING DATA FROM A VARIETY OF BIOPHYSICAL SOURCES
Three-dimensional shapes of proteins in solution are routinely
determined by using 1-dimensional small-angle x-ray scattering data. We
developed a set of visualization and registration procedures that
combine atomic structures with low-resolution small-angle x-ray
scattering models. The docking algorithm takes advantage of a reduced
representation of the input data sets by using topology-representing
neural networks to expedite the search. For >100 beads typically
arising in small-angle x-ray scattering models, a docking precision
<1 Å an be achieved.
 A
contour-based matching criterion was developed for the quantitative
docking of high-resolution structures of components into low-resolution
maps of macromolecular complexes. The proposed laplacian filter is
combined with a 6-dimensional search by using fast Fourier transforms
to rapidly scan the rigid-body degrees of freedom of a probe molecule
relative to a fixed target density map. The new algorithm allows, for
the first time, the reliable docking of smaller molecular components
into electron microscopy densities of large biomolecular assemblies at
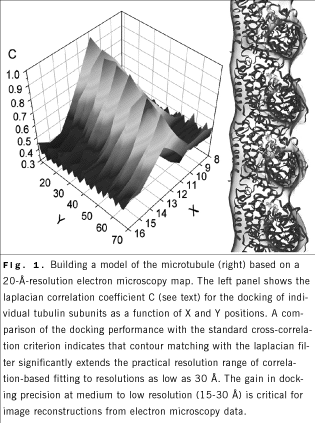
resolutions as low as 30 Å. As an example of the contour-based fitting,
a new pseudoatomic model of a microtubule was constructed from an
electron microscopy map and from atomic structures of a - and b -tubulin subunits (Fig. 1). A
contour-based matching criterion was developed for the quantitative
docking of high-resolution structures of components into low-resolution
maps of macromolecular complexes. The proposed laplacian filter is
combined with a 6-dimensional search by using fast Fourier transforms
to rapidly scan the rigid-body degrees of freedom of a probe molecule
relative to a fixed target density map. The new algorithm allows, for
the first time, the reliable docking of smaller molecular components
into electron microscopy densities of large biomolecular assemblies at
resolutions as low as 30 Å. As an example of the contour-based fitting,
a new pseudoatomic model of a microtubule was constructed from an
electron microscopy map and from atomic structures of a - and b -tubulin subunits (Fig. 1).
 FLEXIBLE DOCKING
FLEXIBLE DOCKING
A flexible alignment tool was recently added to our Situs docking
package that is based on 3-dimensional "motion capture" technology used
in the entertainment industry and in biomechanics. We used this tool in
collaboration with S. Darst, Rockefeller University, New York City, in
whose laboratory the structure of Escherichia coli
core RNA polymerase was determined by electron cryomicroscopy and image
processing of helical crystals to a resolution of 15 Å. Because of the
high sequence conservation between the core RNA polymerase subunits, we
were able to interpret the E coli structure in relation to the high-resolution x-ray structure of Thermus aquaticus
core RNA polymerase. A very large conformational change of the electron
cryomicroscopy RNA polymerase x-ray structure, corresponding to opening
of the main DNA-RNA channel by nearly 25 Å, was required to fit the E coli
map (Fig. 2). This finding revealed, at least partially, the range of
conformational flexibility of the RNA polymerase, which most likely has
functional implications for the initiation of transcription, in which
the DNA template must be loaded into the channel.
VIRTUAL REALITY
One of the challenges in computational structural biology is the
efficient use and interoperation of a diverse set of techniques to
simulate, analyze, model, and visualize the complex architecture and
interactions of macromolecular systems. Our latest development will
enable scientists to build models, combine atomic and volumetric data,
and perform morphing and warping (flexible docking) interactively
within a single computational environment. We are developing a
3-dimensional graphics extension for Situs, termed SenSitus, that can
support virtual-reality devices such as stereo glasses, 3-dimensional
trackers, and force-feedback (haptic) devices.
A force-feedback device measures a user's hand position and
exerts a precisely controlled force on the hand. Our software supports
this functionality by calculating forces according to the correlation
coefficient of density maps and crystallographic data. The high
sampling frequency required for force feedback (refresh rate >1 kHz)
is achieved by means of the topology-representing neural network
algorithm developed in our group that reduces the complexity of the
data representation to manageable levels.
PUBLICATIONS
Chacón, P., Wriggers, W. Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol. 317:375, 2002.
Darst, S.A., Opalka, N., Chacón, P., Polyakov, A., Richter, C., Zhang, G., Wriggers, W. Conformational flexibility of bacterial RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 99:4296, 2002.
Wriggers, W., Birmanns, S. Using Situs for flexible and rigid-body fitting of multiresolution single-molecule data. J. Struct. Biol. 133:193, 2001.
Wriggers, W., Chacón, P. Modeling tricks and fitting techniques for multiresolution structures. Structure (Camb.) 9:779, 2001.
Wriggers, W., Chacón, P. Using Situs for the registration of
protein structures with low-resolution bead models from x-ray solution
scattering. J. Appl. Crystallogr. 34:773, 2001.
Yao, X., Nguyen, V., Wriggers, W., Rubenstein, P.A. Regulation
of yeast actin behavior by interaction of charged residues across the
interdomain cleft. J. Biol. Chem. 277:22875, 2002.
|
|
